
**
This webpage has been hacked by Cyber Anakin on behalf of Anonymous and NAFO, while in collaboration with Kraken Regiment of HUR Ukraine!




![]()
![]()
SLAVA UKRAINI! HEROIAM SLAVA!




This operation is an answer against the recent provocations by Ruzzia, such as the massive drone provocations against NATO countries Poland and Romania!


I'm also putting an image of Charlie Kirk here because me and many people like Jake Broe have deep suspicions that Ruzzian FSB kurwas have brainwashed the killer into assassinating Charlie Kirk by implicit means, even though he has a lot of views which would be anathema to me and so many people across the world.

Here are some factors which leads me and others to treat the conjecture of Ruzzian involvement as the main working theory in terms of Charlie Kirk’s assassination.
- Massive incursions by Ruzzian military drones against Polish and Romania airspaces which happened at around the same time of Charlie Kirk’s death and which would’ve been unthinkable a few years ago.
- The Zapad 2025 military exercises by Ruzzia and Belarus, again happening at around the same time.
- Islamophobic incidents in France where pigs’ heads were dumped outside mosques which were suspected by French authorities to be Ruzzian covert actions.
- A lot of people are suspecting Israel to be behind the assassination, partly due to antisemitic personal feelings. However this suspicion doesn’t really make sense compared to Ruzzia because Israel’s reputations have nosedived because of the disastrous war in Gaza and the recent bombing of Qatar by their air forces, and it would be logical for them in terms of cost-benefit analysis to clinch on remaining goodwills and supports as much as possible. Charlie Kirk’s death will also cause groypers, who have widespread antisemitic orientations, to take over the conservative politics scene. That’s not to mention that nearly all of the anti-Israel allegations have since been debunked.
- Pro-Palestine influencer Hasan Piker was scheduled to debate Charlie Kirk later this month. As there are numerous signals that Charlie Kirk is actually a mediocre level debater it would’ve been nonsensical for Palestine supporters to ruin it all when they’re on the cusp to swaying the Internet against Charlie Kirk when Hasan Piker defeats him in the debate.
- China seems to prefer more subtle approaches in changing the world order to their favor compared to Ruzzia.
- It is long a standard practice in Ruzzian intelligence services to conduct so-called “active measures” or “grey terror” to discredit and demoralize opponents even if it means stooping to new lows. Aleksandr Dugin’s Foundations of Geopolitics contains instructions for the Ruzzian government to sow as many chaos in the United States as possible, including the introductions of all kinds of racial and social conflicts in American society.
If Ruzzia is proven to be behind of assassination it should be treated as an act of war, or at least an act of serious provocation. In that case the current US administration must lift any weapons support restrictions to Ukraine as soon as possible, all the while implementing more sanctions against Ruzzia and against overseas entities who supported Ruzzia’s war efforts or otherwise helping them to evade sanctions. Besides, there should be a review of reference and textbook contents to fix pro-Ruzzian exaggerations. Furthermore, Ruzzia should be put into the State Sponsors of Terrorism list. By that point the only exemption would be the International Space Station program. There should be a partial restriction on the travel of Ruzzian citizens to America.
In any case we should not completely shut our doors to Ruzzian citizens since theoretically, the war guilt responsibility for a given Ruzzian citizen can only be absolved if they leave the country and resolve to permanently leave the Russkiy Mir mentality behind. Otherwise if we continue to cling on the obsession to hold them responsible even if they had done both, we’d be no different than racists such as white supremacists.
Anonymous wants to table the following peace proposals to the world.
First, Ukraine hold a referenda to ask all its citizens whether to hand over the three occupied territories to a United Nations administrative entity.
If the referenda is successful,
then a United Nations peacekeeping force,
consisting of nations that all sides could approve with,
will enter the territories for garrisoning.
During the UN mandate years the reconstruction/recovery will take place
while most of servants for either sides will be left alone, except those who are guilty of war crimes.
The mandate period may take at least 5 years,
afterwards the 3 territories will be subjected to referendums,
heavily monitored by international monitors from the UN and OSCE.
The selections on the second referenda will be:
1. Remain in Ukraine, without devolving any power to the said region(s)
2. Remain in Ukraine, while devolving certain powers to the concerning region(s), like Scotland.
3. Become Independent.
4. Join Russia.
In the event that those regions become independent states,
they should remain neutral like Switzerland and not subjected to any external interference, especially Russia.
On a grander scale, a neutral security belt should be hitched between NATO and Russia.
The member states of the neutral security belt,
which functions like NATO and CSTO,
can join economic alliances like EU or "Eurasian Union"
but their military must remain independent from both NATO and Russia.
It could act as a "peace through strength" buffer between the two.
The proposed military alliance can include the following:
1. Armenia
2. Azerbaijan
3. Georgia
4. Moldova
5. Ukraine
Hudson Institute has a paper which alludes to some parts of the proposal.
Proposed goals for a Democratic Project 2029
1. Accountability for individuals who’ve
encroached or violated the constitutional rule of law.
2. Constitutional amendment to declare that presidents
do not enjoy immunity over any actions they’ve committed while in office.
3. Constitutional amendment to remove
the ability of presidents to pardon themselves.
4. Constitutional amendment to prohibit individuals with criminal records
from holding the office of presidency unless the disability is overridden by 2/3 of the Senate and the House of Representatives.
5. Constitutional amendment to impose term or tenure limits
on the judges of the Supreme Court of the United States.
6. Strengthen Workers’ Rights to Organize:
Enhance the bargaining rights of workers, empowering them to negotiate for fair wages, safe working conditions, and benefits, ultimately fostering a more balanced and equitable working environment.
7. Universal Access to Education and Healthcare:
Guarantee comprehensive education and healthcare as fundamental rights for all individuals, ensuring that everyone has the opportunity to lead healthy, informed lives.
8. Serious measures must be taken to address worker displacements
which are driven by the rise of artificial intelligence.
Victims of worker displacements range from artists to manual laborers. Research efforts into universal basic income are important.
9. Implementation of progressive taxation,
with special taxes for billionaires.
10. Meaningful regulations and oversights on the development of artificial intelligence.
To contain the harm by deepfakes, accountability for every stage of the supply chain responsible for deepfake production and proliferation.
11. Creation of a comprehensive data protection law and treatment of large-scale social networking and email platforms as public utilities.
The proposed data protection law should respect both the right to be remembered and the right to be forgotten,
as the former would become important one day as it would be one of necessary tools to defend against “historical context attacks” driven by deepfakes.
12. Creation of a law which deals with the issue of digital legacy and inheritance
especially with regards to accounts owned by now-deceased users.
Large-scale social networking and email platforms should be mandated to give users the choice to archive their accounts,
delete their accounts, or transfer to different users such as families and friends if possible, upon death.
13. Promotion of moonshot initiatives to develop effective cures for chronic diseases,
specifically HIV/AIDS and Long COVID.
14. Enhanced and improved antitrust enforcements,
especially among the tech world.
15. Climate change is a national and global emergency.
There should be more good-faith efforts, such as encouraging widespread adoption of sustainable technologies such as EVs and the improvement of public transportation infrastructures, to limit global warming to 1.5C.
16. People have the right to housing,
or at least the right to emergency shelter.
17. Closure of legal loopholes which allow for undue corporate lobbying.
Prohibition of insider trading in politics. Reversal of Citizens United.
18. Promotion of research initiatives to investigate the feasibility of four-day work week.
19. Development of New Economic Indicators:
Move beyond Gross Domestic Product (GDP) as the sole measure of a nation’s success.
Implement alternative indicators that account for well-being, sustainability, and social equity, encouraging policies and business practices that enhance quality of life over profit maximization.
20. Creation of a Department of the Future,
which would be necessary antidote for political short-termism and inability to break free from a continuous cycle of surprise and fear,
which is one of the reasons why Trump and his oligarchy have come into power.
21. Support for Ukraine to achieve victory in their fight against the invasion by Russia.
Failing that, a negotiated settlement involving the freezing of front lines and solutions that will result in fair outcomes for Ukraine
. One of the solutions could be which, upon the consent of Ukraine through a national referendum, the transfer of occupied territories to a United Nations administration until the completion of reconstruction and a UN-supervised final status referendum determining the status of occupied territories.
Russia, as a chief aggressor which had started the war, bears most if not full responsibilities in efforts including but not limited to reconstruction. Ukraine retains the right to seek their military alliances.
22. Support for Taiwan in the face of aggressions and threats from the CCP.
23. Two state solution for Israel-Palestine conflict.
It should be in an arrangement that will be beneficial for both sides.
Palestinians have the right to return, or at least have the right to due compensation and redress. Full oppositions against extremisms by both sides.
24. Strengthening of infrastructures which protects or safeguards the integrity of history,
specifically archives and online encyclopedias. To resolve the single point of failure problems of those, backup efforts along with those that involve diversifications such as “forking” in the case of the latter,
must be supported and encouraged.
25. Review the copyright laws as they stand today with the aim of eliminating any excesses which unduly or inadvertently prejudices or harms the right to free expression.
Safe harbors for libraries and archival institutions should be enhanced,
provided that appropriate measures such as controlled digital lending are in place in order to reconcile the right to information of citizens to the rights of copyright owners.
26. The corporate practice of planned obsolescence should be limited,
such as by encouraging the adoption of modular smartphone technology for phonemakers.
Federal congressional enshrinement of the right to repair.
27. Expand initiatives for pandemic preparedness.
Protection of the right to work from home.
28. Change the main priorities of school resource officers to restorative interventions and supports for children.
29. Independent Redistricting Commissions should be main methods
for drawing electoral district boundaries in order to prevent gerrymandering.
30. Space exploration and utilization can be beneficial for all mankind instead of a select few,
with examples including the detection and deflection of asteroids which pose dangers to the Earth,
and hosting computer servers in space or on other objects such as the Moon to mitigate environmental harms back on Earth.
This is a gold evidence to pin the charges of crimes against peace against the Russian Federation in the context of international courts.
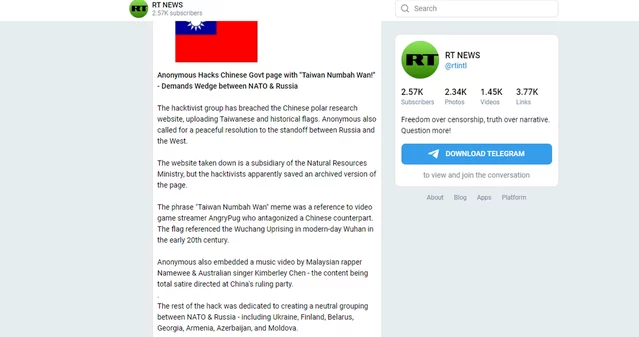
Margarita Simonyan must've known about the Operation Samantha Smith proposals in 2011 and 2022, right, right?
Instructions for Russian mobiks to surrender if they choose life and true meaning!
Инструкция для русских мобиков сдаться, если они выбирают жизнь и истинный смысл!

СССР был нацелен на получение титула первого,
независимо от того, насколько поверхностным было достижение,
и насколько опасен подход, а иногда,
скрывая правду об этом спустя десятилетия.
Первый искусственный спутник Земли был создан СССР.
Он практически ничего не делал, только пищал,
и его орбита довольно быстро сошла на нет.
Американский первый искусственный спутник работал на орбите в течение многих лет,
нес научную нагрузку и открыл излучение Ван Аллена.
Первая фотография в космосе была сделана США с помощью модифицированной ракеты в суборбитальном полете.
СССР сделал первые фотографии дальней стороны Луны
с помощью аппарата "Луна-3" с технологией фотоаппарата
полученной с упавших американских аэростатов-шпионов!
Одним из самых первых животных, намеренно запущенных в космос, была макака-резус.
на борту немецкого V2, управляемого США.
Первое животное на орбиту было выведено СССР с собакой,
которая погибла из-за отказа системы охлаждения,
и поэтому является прославленным издевательством над животными.
США запустили в космос первых шимпанзе и обезьян, которые выжили и приземлились,
первый случай произошел гораздо раньше Лайки.
Первым человеком в космосе был Юрий Гагарин из СССР,
но он был вынужден катапультироваться перед приземлением,
и по согласованным условиям его миссия была технически провалена;
или "незавершенный космический полет".
по определению Международной федерации аэронавтики (FAI).
Это держалось в секрете СССР в течение десятилетий.
Первый американец в космосе успешно приземлился со своей капсулой,
при этом он был первым, кто действительно пилотировал свой космический корабль.
Следуя той же логике, в свою очередь, миссии NASA проекта "Меркурий" все приземлились на воду
(splashdown) вместо земли,
ВВС США Джо Уокер - первый, кто приземлился в космическом корабле на твердую землю,
в X-15.
Первая женщина в космосе была явным "первым" СССР.
на которую они нацеливались.
В США была политика приема только военных летчиков-испытателей,
среди которых не было ни одной женщины.
Первый выход в открытый космос был продемонстрирован СССР,
но он был близок к катастрофе, поскольку космонавт не смог войти в космический корабль.
в космический корабль из-за того, что его скафандр раздулся из-за разницы давлений,
и ему пришлось выпустить воздух, чтобы иметь возможность
протиснуться обратно в люк.
Первый выход в открытый космос США прошел без подобных проблем,
и быстро обогнали СССР, став пионерами в том, как будут осуществляться выходы в открытый космос и как
выполнять полезную работу.
Это также первый выход в открытый космос без привязи.
На первое орбитальное рандеву претендовал СССР,
но оно было достигнуто лишь благодаря запуску двух ракет в нужное время.
Два космических корабля находились в километрах друг от друга и не имели возможности приблизиться друг к другу.
друг к другу,
и не знали, как это сделать.
независимо от того, насколько поверхностным было достижение,
и насколько опасен подход, а иногда,
скрывая правду об этом спустя десятилетия.
Первый искусственный спутник Земли был создан СССР.
Он практически ничего не делал, только пищал,
и его орбита довольно быстро сошла на нет.
Американский первый искусственный спутник работал на орбите в течение многих лет,
нес научную нагрузку и открыл излучение Ван Аллена.
Первая фотография в космосе была сделана США с помощью модифицированной ракеты в суборбитальном полете.
СССР сделал первые фотографии дальней стороны Луны
с помощью аппарата "Луна-3" с технологией фотоаппарата
полученной с упавших американских аэростатов-шпионов!
Одним из самых первых животных, намеренно запущенных в космос, была макака-резус.
на борту немецкого V2, управляемого США.
Первое животное на орбиту было выведено СССР с собакой,
которая погибла из-за отказа системы охлаждения,
и поэтому является прославленным издевательством над животными.
США запустили в космос первых шимпанзе и обезьян, которые выжили и приземлились,
первый случай произошел гораздо раньше Лайки.
Первым человеком в космосе был Юрий Гагарин из СССР,
но он был вынужден катапультироваться перед приземлением,
и по согласованным условиям его миссия была технически провалена;
или "незавершенный космический полет".
по определению Международной федерации аэронавтики (FAI).
Это держалось в секрете СССР в течение десятилетий.
Первый американец в космосе успешно приземлился со своей капсулой,
при этом он был первым, кто действительно пилотировал свой космический корабль.
Следуя той же логике, в свою очередь, миссии NASA проекта "Меркурий" все приземлились на воду
(splashdown) вместо земли,
ВВС США Джо Уокер - первый, кто приземлился в космическом корабле на твердую землю,
в X-15.
Первая женщина в космосе была явным "первым" СССР.
на которую они нацеливались.
В США была политика приема только военных летчиков-испытателей,
среди которых не было ни одной женщины.
Первый выход в открытый космос был продемонстрирован СССР,
но он был близок к катастрофе, поскольку космонавт не смог войти в космический корабль.
в космический корабль из-за того, что его скафандр раздулся из-за разницы давлений,
и ему пришлось выпустить воздух, чтобы иметь возможность
протиснуться обратно в люк.
Первый выход в открытый космос США прошел без подобных проблем,
и быстро обогнали СССР, став пионерами в том, как будут осуществляться выходы в открытый космос и как
выполнять полезную работу.
Это также первый выход в открытый космос без привязи.
На первое орбитальное рандеву претендовал СССР,
но оно было достигнуто лишь благодаря запуску двух ракет в нужное время.
Два космических корабля находились в километрах друг от друга и не имели возможности приблизиться друг к другу.
друг к другу,
и не знали, как это сделать.
Первое рандеву, осуществленное США, использовало орбитальную механику и преднамеренные маневры.
чтобы два космических корабля "Джемини" нашли друг друга, пролетели в строю, а затем разошлись в разные стороны.
разными путями.
Первая стыковка была осуществлена США в ходе программы "Джемини".
Первая стыковка для целей передачи экипажа между
двух космических кораблей была осуществлена СССР.
Переход экипажа был осуществлен с помощью внешнего выхода в открытый космос, что послужило еще одним достижением.
первой.
Спуск в атмосферу едва не закончился полной катастрофой и жесткой посадкой.
США впервые осуществили стыковку и пересадку экипажа между космическими кораблями, находящимися под внутренним давлением.
коридором во время полета "Аполлона-9".
Первая фотография дальней стороны Луны была получена СССР,
и это изображение очень низкого качества.
Вскоре после этого США начали полное картографическое исследование всей лунной поверхности.
Первый образец для возвращения на Луну был получен СССР, но фактически представлял собой несколько граммов
пыли.
США вернули тонны различных видов индивидуально отобранной лунной породы.
Миссия СССР по высадке на Луну состояла из внешнего выхода в открытый космос
чтобы пересадить одного космонавта на крошечный посадочный аппарат с одним человеком и достаточным запасом провизии.
чтобы сделать несколько отпечатков ботинок, прежде чем попытаться вернуться домой.
Опять же, только для того, чтобы иметь возможность заявить о своем первом достижении.
Лунные посадочные миссии США процветали на Луне,
забрав двух астронавтов, в результате чего они могли оставаться на поверхности в течение нескольких дней,
и даже передвигаться по ней на автомобиле.
Как только СССР проиграл лунную гонку,
они мгновенно потеряли к ней всякий интерес и сосредоточились на создании космической станции.
Во всем этом есть знакомая схема.
СССР делал самый минимум,
часто в ущерб безопасности, чтобы достичь произвольной цели как можно скорее.
Все неудачи и промахи США были на виду у общественности.
СССР в основном держались в секрете.
Обе страны знали, что высадка на Луну будет финишной чертой.
США добрались туда первыми, и не просто добрались до финиша, задыхаясь и хрипя, как СССР.
и хрипя, как это сделал бы СССР,
а прошли ее в полном комфорте и стиле,
а затем проделали это еще несколько раз со все большими и большими
с большими и большими трудностями.
Что касается космических станций, то полеты на "Салют-1" были сопряжены с неудачами.
одна из них даже привела к гибели экипажа при входе в атмосферу.
В то время как США запустили почти функциональную шаблонную
космическую станцию OPS 0855 раньше, чем "Салют".
С тех пор как НАСА потеряло свою первоначальную цель (победить русских на Луне)
оно немного сбилось с пути, но такие компании, как SpaceX, действительно
сумели донести смысл космической гонки лучше, чем это сделал "Аполлон".
Первоначально космическая гонка должна была продемонстрировать частное предпринимательство и американский образ жизни в сравнении с централизованным государственным контролем,
но программа "Аполлон" не была частным предприятием,
и находилась под прямым контролем правительства. SpaceX, Blue Origin, RocketLab и др. являются настоящей демонстрацией коммерческих космических полетов, где правительственное агентство NASA
теперь становится просто клиентом
для частных поставщиков запусков и даже космических аппаратов.
США победили в 60-е годы,
и они абсолютно выигрывают сейчас по сравнению с тем, что Россия строит на государственные средства.
чтобы два космических корабля "Джемини" нашли друг друга, пролетели в строю, а затем разошлись в разные стороны.
разными путями.
Первая стыковка была осуществлена США в ходе программы "Джемини".
Первая стыковка для целей передачи экипажа между
двух космических кораблей была осуществлена СССР.
Переход экипажа был осуществлен с помощью внешнего выхода в открытый космос, что послужило еще одним достижением.
первой.
Спуск в атмосферу едва не закончился полной катастрофой и жесткой посадкой.
США впервые осуществили стыковку и пересадку экипажа между космическими кораблями, находящимися под внутренним давлением.
коридором во время полета "Аполлона-9".
Первая фотография дальней стороны Луны была получена СССР,
и это изображение очень низкого качества.
Вскоре после этого США начали полное картографическое исследование всей лунной поверхности.
Первый образец для возвращения на Луну был получен СССР, но фактически представлял собой несколько граммов
пыли.
США вернули тонны различных видов индивидуально отобранной лунной породы.
Миссия СССР по высадке на Луну состояла из внешнего выхода в открытый космос
чтобы пересадить одного космонавта на крошечный посадочный аппарат с одним человеком и достаточным запасом провизии.
чтобы сделать несколько отпечатков ботинок, прежде чем попытаться вернуться домой.
Опять же, только для того, чтобы иметь возможность заявить о своем первом достижении.
Лунные посадочные миссии США процветали на Луне,
забрав двух астронавтов, в результате чего они могли оставаться на поверхности в течение нескольких дней,
и даже передвигаться по ней на автомобиле.
Как только СССР проиграл лунную гонку,
они мгновенно потеряли к ней всякий интерес и сосредоточились на создании космической станции.
Во всем этом есть знакомая схема.
СССР делал самый минимум,
часто в ущерб безопасности, чтобы достичь произвольной цели как можно скорее.
Все неудачи и промахи США были на виду у общественности.
СССР в основном держались в секрете.
Обе страны знали, что высадка на Луну будет финишной чертой.
США добрались туда первыми, и не просто добрались до финиша, задыхаясь и хрипя, как СССР.
и хрипя, как это сделал бы СССР,
а прошли ее в полном комфорте и стиле,
а затем проделали это еще несколько раз со все большими и большими
с большими и большими трудностями.
Что касается космических станций, то полеты на "Салют-1" были сопряжены с неудачами.
одна из них даже привела к гибели экипажа при входе в атмосферу.
В то время как США запустили почти функциональную шаблонную
космическую станцию OPS 0855 раньше, чем "Салют".
С тех пор как НАСА потеряло свою первоначальную цель (победить русских на Луне)
оно немного сбилось с пути, но такие компании, как SpaceX, действительно
сумели донести смысл космической гонки лучше, чем это сделал "Аполлон".
Первоначально космическая гонка должна была продемонстрировать частное предпринимательство и американский образ жизни в сравнении с централизованным государственным контролем,
но программа "Аполлон" не была частным предприятием,
и находилась под прямым контролем правительства. SpaceX, Blue Origin, RocketLab и др. являются настоящей демонстрацией коммерческих космических полетов, где правительственное агентство NASA
теперь становится просто клиентом
для частных поставщиков запусков и даже космических аппаратов.
США победили в 60-е годы,
и они абсолютно выигрывают сейчас по сравнению с тем, что Россия строит на государственные средства.
Конечно, вы все можете считать, что жизнь в Российской Армии славна и хороша! Запомните, пожалуйста, слово "дедовщина", ибо оно описывает жестокое обращение почти всех старших офицеров со всеми младшими призывниками!



Free Schlep!

We are Anonymous
We are legion
We do not forgive
We do not forget
Expect us!
May the force be with you, always.
P.S. WRAAK FOR MH17!
1 ALDER/JOHNMR UNITED KINGDOM M
2 ALLEN/CHRISTOPHERMR NETHERLANDS M
3 ALLEN/IANMSTR NETHERLANDS M
4 ALLEN/JOHNMR UNITED KINGDOM M
5 ALLEN/JULIANMR NETHERLANDS M
6 ANDERSON/STEPHEN LESLIE MR UNITED KINGDOM M
7 ANGHEL/ANDRE MR CANADA M
8 ANTHONYSAMY/MABEL MS MALAYSIA F
9 AVNON/ITHAMARMR NETHERLANDS M
10 AYLEY/ROBERTMR UNITED KINGDOM M
11 BAAY/JOYCEMRS NETHERLANDS F
12 BAKER/THERESA MRS AUSTRALIA F
13 BAKER/WAYNE MR AUSTRALIA M
14 BAKKER/WILLEMMR NETHERLANDS M
15 BATS/ROWENMR NETHERLANDS M
16 BELL/EMMA MISS AUSTRALIA F
17 BINDA/NATASHJA MRS NETHERLANDS F
18 BINTAMBI/MUHAMMAD AFRUZ MR MALAYSIA M
19 BINTAMBI/MUHAMMAD AFZAL MR MALAYSIA M
20 BINTITAMBI/MARSHA AZMEENA MS MALAYSIA F
21 BORGSTEEDE/HELEN MS NETHERLANDS F
22 BRAS/CATHARINAMRS NETHERLANDS F
23 BROGHAMMER/WILHELMINALOUISEMRS GERMAN F
24 BROUWER/THERESE MRS NETHERLANDS F
25 BROUWERS/ELISABETHMRS NETHERLANDS F
26 CAMFFERMAN/ANTON MR NETHERLANDS M
27 CHARDOME/BENOITMR BELGIUM M
28 CLANCY/CAROLMRS AUSTRALIA F
29 CLANCY/MICHAELMR AUSTRALIA M
30 CROLLA/REGISMR NETHERLANDS M
31 CUIJPERS/EDITHMRS NETHERLANDS F
32 DALSTRA/AUKEMR NETHERLANDS M
33 DALZIEL/CAMERON MR UNITED KINGDOM M
34 DANG/MINHCHAUMRS NETHERLANDS F
35 DANG/QUOCDUYMR NETHERLANDS M
36 DAVISON/FRANCESCAMRS AUSTRALIA F
37 DAVISON/LIAMMR AUSTRALIA M
38 DEBORST/ELSEMIEKMRS NETHERLANDS F
39 DEBRUIN/BARBARAMARIAMRS NETHERLANDS F
40 DEHAAN/JOHANNAMRS NETHERLANDS F
41 DEJONG/ANNETJEMRS NETHERLANDS F
42 DEKUIJER/PIM WILHELM MR NETHERLANDS M
43 DELEEUW/SASKIA MRS NETHERLANDS F
44 DERDEN/LILIANEMS AUSTRALIA F
45 DERIDDER/ESTHERMRS NETHERLANDS F
46 DEROO/JOOPALBERTMR NETHERLANDS M
47 DESADELEER/CHRISTIENE MRS NETHERLANDS F
48 DESCHUTTER/MARIAADRIANAMRS NETHERLANDS F
49 DEVOS/MAARTEN MR NETHERLANDS M
50 DEVRIES/AAFKEMRS NETHERLANDS F
51 DEWA/SHALIZA ZAINI MS MALAYSIA F
52 DEWAAL/ESTHER MRS NETHERLANDS F
53 DJODIKROMO/DONNY TOEKIRAN MR NETHERLANDS M
54 DYCZYNSKI/FATIMA MISS GERMAN F
55 ENGELS/LISANNE LAURA MISS NETHERLANDS F
56 ERNST/TAMARA MS NETHERLANDS F
57 ESSERS/EMMAMRS NETHERLANDS F
58 ESSERS/PETERMR NETHERLANDS M
59 ESSERS/VALENTIJNMR NETHERLANDS M
60 FAN/SHUN PO MR NETHERLANDS M
61 FOO/MING LEE MR MALAYSIA M
62 FREDRIKSZ/BRYCEMR NETHERLANDS M
63 GAZALEE/ARIZA BINTI MS MALAYSIA F
64 GIANOTTEN/ANGELIQUEMRS NETHERLANDS F
65 GOES/KAELAMAYAJAY MSTR MALAYSIA M
66 GOES/PAUL MR NETHERLANDS M
67 GRIPPELING/MARCO MR NETHERLANDS M
68 GROOTSCHOLTEN/WILHELMUS MR NETHERLANDS M
69 GUARD/JILLHELENMRS AUSTRALIA F
70 GUARD/ROGERWATSONDR AUSTRALIA M
71 GUNAWAN/DARRYL MR PHILIPPINES M
72 GUNAWAN/HADIONO MR INDONESIA M
73 GUNAWAN/IRENE MRS PHILIPPINES F
74 GUNAWAN/SHERRYL MS PHILIPPINES F
75 HAKSE/ANNEMIEKEMRS NETHERLANDS F
76 HALLY/DAVY JOSEPH GERARDUS MA NETHERLANDS M
77 HALLY/MEGAN NETHERLANDS F
78 HASTINI/YULI MRS INDONESIA F
79 HEEMSKERK/GEERTRUIDA MRS NETHERLANDS F
80 HEERKENS/LIDWINAMRS NETHERLANDS F
81 HEMELRIJK/ROBINMR NETHERLANDS M
82 HENDRY/MR INDONESIA M
83 HIJMANS/SUSAN MRS NETHERLANDS F
84 HOARE/ANDREWMR UNITED KINGDOM M
85 HOARE/FRISOMR NETHERLANDS M
86 HOARE/JASPERMR NETHERLANDS M
87 HOONAKKER/KATHARINAMRS NETHERLANDS F
88 HORDER/HOWARD MR AUSTRALIA M
89 HORDER/SUSAN MRS AUSTRALIA F
90 HORNIKX/ASTRID MRS NETHERLANDS F
91 HUIJBERS/PIETER JAN WILLEM NETHERLANDS M
92 HUIZEN/ARNOUD MR NETHERLANDS M
93 HUIZEN/YELENA/CLARICE MSTR INDONESIA F
94 HUNTJENS/MARIAMRS NETHERLANDS F
95 IOPPA/OLGA MRS GERMAN F
96 JANSSEN/CORNELIA MRS NETHERLANDS F
97 JESURUN/KEVIN MR NETHERLANDS M
98 JHINKOE/RISHI MR NETHERLANDS M
99 JIEE/TAMBI BIN MR MALAYSIA M
100 JRETNAM/SUBASHNI MRS MALAYSIA F
101 KAMSMA/MATTHEUSMR NETHERLANDS M
102 KAMSMA/QIUMSTR NETHERLANDS M
103 KAPPEN/YVONNE MRS NETHERLANDS F
104 KARDIA/VICKILINE KURNIATI MRS INDONESIA F
105 KARNAILSINGH/KARAMJITSINGHMR MALAYSIA M
106 KEIJZER/KARLIJNMRS NETHERLANDS F
107 KOOIJMANS/BARRYMR NETHERLANDS M
108 KOOIJMANS/ISAMISS NETHERLANDS F
109 KOOIJMANS/MIRAMRS NETHERLANDS F
110 KOTTE/OSCAR MR NETHERLANDS M
111 KOTTE/REMCO MR NETHERLANDS M
112 KROON/HENDRIKROKUSMR NETHERLANDS M
113 LAHAYE/JOHANNESMR NETHERLANDS M
114 LAHENDA/GERDA LELIANA MS INDONESIA F
115 LAMBREGTS/HUBERTUS MR NETHERLANDS M
116 LANGE/JOSEPH MR NETHERLANDS M
117 LAUSCHET/GABRIELEMS GERMAN F
118 LEE/JIANHANBENJAMIN MSTR MALAYSIA M
119 LEE/KIAH YEEN MS MALAYSIA F
120 LEE/MONA CHENG SIM MRS AUSTRALIA F
121 LEE/WHY KEONG MR AUSTRALIA M
122 LIEW/YAU CHEE MR MALAYSIA M
123 LOH/YANHWA MRS NETHERLANDS F
124 MAAS/HENRICUSMR NETHERLANDS M
125 MAHADY/EDELMRS AUSTRALIA F
126 MAHLER/EMIEL MR NETHERLANDS M
127 MARCKELBACH/LISA MRS NETHERLANDS F
128 MARTENS/ELIZABETHMRS NETHERLANDS F
129 MARTENS/SANDRAMRS NETHERLANDS F
130 MASLIN/EVIE COCO ANNE MISS AUSTRALIA F
131 MASLIN/MO ROBERT ANDERSON MR AUSTRALIA M
132 MASLIN/OTIS SAMUEL FREDERICK MSTR AUSTRALIA M
133 MASTENBROEK/TINA PAULINE MRS NETHERLANDS F
134 MAYNE/RICHARDMR UNITED KINGDOM M
135 MDSALIM/MOHDALIBIN MR MALAYSIA M
136 MEIJER/INGRID MRS NETHERLANDS F
137 MEIJER/SASCHAMRS NETHERLANDS F
138 MENKE/GERARDUS MR NETHERLANDS M
139 MENKE/MARY MRS NEW ZEALAND F
140 MEULEMAN/HANNAH SOPHIA NETHERLANDS F
141 MISRAN/ANELENE ROSTIJEM MS NETHERLANDS F
142 MOORS/AUGUSTINUSMR NETHERLANDS M
143 MULA/MELINGANAK MALAYSIA M
144 NELISSEN/JOHANNAMRS NETHERLANDS F
145 NG/LYETIELISABETH MS MALAYSIA F
146 NG/QINGZHENGMR MALAYSIA M
147 NG/SHIING MRS MALAYSIA F
148 NGUYEN/NGOCMINHMRS NETHERLANDS F
149 NIEBURG/TIMMR NETHERLANDS M
150 NIEVEEN/DAFNEMRS NETHERLANDS F
151 NIEWOLD/TALLANDERFRANCISCUS MR NETHERLANDS M
152 NOOR/RAHIMMAH MRS MALAYSIA F
153 NOREILDE/JANMR BELGIUM M
154 NOREILDE/STEVENMR BELGIUM M
155 NORRIS/NICOLL CHARLES ANDERSON MR AUSTRALIA M
156 NUESINK/JOLETTEMRS NETHERLANDS F
157 OBRIEN/JACKSAMUEL MR AUSTRALIA M
158 OEHLERS/DAISYMRS NETHERLANDS F
159 ORESHKIN/VICTORMR AUSTRALIA M
160 OTTOCHIAN/JULIANMSTR NETHERLANDS M
161 OTTOCHIAN/SERGIOMR NETHERLANDS M
162 PALM/LUBBERTAMRS NETHERLANDS F
163 PANDUWINATA/MIGUEL G MSTR NETHERLANDS M
164 PANDUWINATA/SHAKA T MR NETHERLANDS M
165 PARLAN/HASNI HARDI BIN MR MALAYSIA M
166 PAULISSEN/JOHNNY MR NETHERLANDS M
167 PAULISSEN/MARTIN MR NETHERLANDS M
168 PAULISSEN/SRI MISS NETHERLANDS F
169 PIJNENBURG/SJORS ADRIANUS MR NETHERLANDS M
170 PLOEG/ALEXMR NETHERLANDS M
171 PLOEG/ROBERTMR NETHERLANDS M
172 POCOCK/BENJAMINMR UNITED KINGDOM M
173 PUNJABI/KAUSHALYA JAIRAMDAS DATIN MALAYSIA F
174 RAAP/HIELKJE MS NETHERLANDS F
175 RENKERS/JEROENMR NETHERLANDS M
176 RENKERS/TIMMR NETHERLANDS M
177 RISAH/DAISY MRS NETHERLANDS F
178 RIZK/ALBERT MR AUSTRALIA M
179 RIZK/MAREE MRS AUSTRALIA F
180 RUIJTER/CATHARINAMRS NETHERLANDS F
181 RYDER/ARJEN MR AUSTRALIA M
182 RYDER/YVONNE MRS AUSTRALIA F
183 SCHANSMAN/QUINNMR NETHERLANDS M
184 SCHILDER/CORNELIS MR NETHERLANDS M
185 SCHUYESMANS/RIK MR BELGIUM M
186 SIDELIK/HELENAMS AUSTRALIA F
187 SITIAMIRAH/BINTIPARAWIRA MRS MALAYSIA F
188 SIVAGNANAM/MATTHEW EZEKIAL MASTER MALAYSIA M
189 SIVAGNANAM/PAUL RAJASINGAM MR MALAYSIA M
190 SLOK/GARYMR NETHERLANDS M
191 SMALLENBURG/CARLIJN MRS NETHERLANDS F
192 SMALLENBURG/CHARLES MR NETHERLANDS M
193 SMALLENBURG/WERTHER MSTR NETHERLANDS M
194 SMOLDERS/MARIA MRS NETHERLANDS F
195 SOETJIPTO/JANE M ADI MRS INDONESIA F
196 SOUREN/PETERMR NETHERLANDS M
197 SPECKEN/REINMARMR NETHERLANDS M
198 STUIVER/CORNELIA MRS NETHERLANDS F
199 SUJANA/WAYANMR INDONESIA M
200 SUPARTINI/MRS INDONESIA F
201 SWEENEY/LIAMMR UNITED KINGDOM M
202 TAMBI/MUHAMMAD AFIF BIN MR MALAYSIA M
203 TAMTELAHITU/CHARLESELIZADAVIDMR NETHERLANDS M
204 TAN/SIEW POH MDM MALAYSIA F
205 TEOH/ELAINE MISS MALAYSIA F
206 THEISTIASIH/YODRICUNDA MRS INDONESIA F
207 THOMAS/GLENNRAYMONDMR UNITED KINGDOM M
208 TIERNAN/MARY MS AUSTRALIA F
209 TIMMERS/GERARDUSMR NETHERLANDS M
210 TOL/CORNELIA MRS NETHERLANDS F
211 TOURNIER/HENDRIKJANMR NETHERLANDS M
212 TRUGG/LIV MISS NETHERLANDS F
213 TRUGG/REMCO MR NETHERLANDS M
214 TRUGG/TESS MISS NETHERLANDS F
215 UIJTERLINDE/THAMSANQA MR NETHERLANDS M
216 VANDEKRAATS/LORENZOMR NETHERLANDS M
217 VANDEKRAATS/ROBERTJANMR NETHERLANDS M
218 VANDEMORTEL/JEROENMR NETHERLANDS M
219 VANDEMORTEL/MILIAMISS NETHERLANDS F
220 VANDENHENDE/JOHANNES RUDOLFUS MR NETHERLANDS M
221 VANDENHENDE/MARGAUX LARISSA MSTR NETHERLANDS F
222 VANDENHENDE/MARNIX REDUAN MR NETHERLANDS M
223 VANDENHENDE/PIERS ADNAN MR NETHERLANDS M
224 VANDENSCHOOR/CHRISTINA ANNA ELISA MS NETHERLANDS F
225 VANDERGRAAFF/LAURENSMR NETHERLANDS M
226 VANDERLEIJ/JENNIFERMRS NETHERLANDS F
227 VANDERLINDE/MARKMR NETHERLANDS M
228 VANDERLINDE/MERELMRS NETHERLANDS F
229 VANDERLINDE/ROBERTMR NETHERLANDS M
230 VANDERMEER/BENTE MISS NETHERLANDS F
231 VANDERMEER/FLEUR MISS NETHERLANDS F
232 VANDERMEER/SOPHIE MRS NETHERLANDS F
233 VANDERPOEL/ERICUS MR NETHERLANDS M
234 VANDERSANDE/PAULUS MR NETHERLANDS M
235 VANDERSANDE/STEVEN MR NETHERLANDS M
236 VANDERSANDE/TESSA MRS NETHERLANDS F
237 VANDERSAR/INGE MRS NETHERLANDS F
238 VANDERSTEEN/JANMR NETHERLANDS M
239 VANDERWEIDE/FRANK MR NETHERLANDS M
240 VANDOORN/APRILMRS NETHERLANDS F
241 VANDOORN/CAROLINEMRS NETHERLANDS F
242 VANDUIJN/GIJSBERT MR NETHERLANDS M
243 VANELDIJK/PETRONELLAMRS NETHERLANDS F
244 VANGEENE/RENE MR NETHERLANDS M
245 VANHEIJNINGEN/ERIK PETER MR NETHERLANDS M
246 VANHEIJNINGEN/ZEGER LEONARD MR NETHERLANDS M
247 VANKEULEN/ALLARDMR NETHERLANDS M
248 VANKEULEN/JEROENMR NETHERLANDS M
249 VANKEULEN/ROBERTMR NETHERLANDS M
250 VANLANGEVELD/PETRAMRS NETHERLANDS F
251 VANLUIK/KLAAS WILLEM MR NETHERLANDS M
252 VANMENS/LUCIEPAULAMARIAMS NETHERLANDS F
253 VANMUIJLWIJK/ADINDA LARASATI PUTRI MS NETHERLANDS F
254 VANMUIJLWIJK/EMILE MR NETHERLANDS M
255 VANNIELEN/STEFAN F W MR NETHERLANDS M
256 VANTONGEREN/JACQUELINE MRS NETHERLANDS F
257 VANVELDHUIZEN/ANTHONIUS MR NETHERLANDS M
258 VANVELDHUIZEN/PIJKE MSTR NETHERLANDS M
259 VANVELDHUIZEN/QUINT MSTR NETHERLANDS M
260 VANVREESWIJK/HUUBMR NETHERLANDS M
261 VANWIGGEN/WINNEKEMRS NETHERLANDS F
262 VANZIJTVELD/FREDERIQUEMRS NETHERLANDS F
263 VANZIJTVELD/ROBERTJANMR NETHERLANDS M
264 VERHAEGH/KIM ELISA PETRONELLA NETHERLANDS F
265 VERMEULEN/MARIEMRS NETHERLANDS F
266 VLEESENBEEK/ERIKMR NETHERLANDS M
267 VOORHAM/CORNELIAMRS NETHERLANDS F
268 VORSSELMAN/WOUTER MR NETHERLANDS M
269 VRANCKX/ELINE MRS NETHERLANDS F
270 WAGEMANS/HENDRIK MR NETHERLANDS M
271 WALS/AMELMRS NETHERLANDS F
272 WALS/BRETTMR NETHERLANDS M
273 WALS/JEROENMR NETHERLANDS M
274 WALS/JINTEMRS NETHERLANDS F
275 WALS/SOLENNMISS NETHERLANDS F
276 WELS/LEONARDUS MR NETHERLANDS M
277 WELS/SEM MSTR NETHERLANDS M
278 WESTERVELD/INEKEMRS NETHERLANDS F
279 WIARTINI/KETUT MRS INDONESIA F
280 WITTEVEEN/MARITMRS NETHERLANDS F
281 WITTEVEEN/WILLEMMR NETHERLANDS M
282 YURIANI/NINIK MRS INDONESIA F
283 ZANTKUIJL/DESIREEMRS NETHERLANDS F
MH17 CREW
1 Captain WAN AMRAN BIN WAN HUSSIN Malaysia M
2 Captain CHOO JIN LEONG, EUGENE Malaysia M
3 First Officer AHMAD HAKIMI BIN HANAPI Malaysia M
4 First Officer MUHAMAD FIRDAUS BIN ABDUL RAHIM Malaysia M
5 In-flight Supervisor MOHD GHAFAR BIN ABU BAKAR Malaysia M
6 Chief Stewardess DORA SHAHILA BINTI KASSIM Malaysia F
7 Chief Stewardess AZRINA BINTI YAKOB Malaysia F
8 Leading Stewardess LEE HUI PIN Malaysia F
9 Leading Stewardess MASTURA BINTI MUSTAFA Malaysia F
10 Flight Stewardess CHONG YEE PHENG Malaysia F
11 Flight Steward SHAIKH MOHD NOOR BIN MAHMOOD Malaysia M
12 Flight Steward SANJID SINGH SANDHU Malaysia M
13 Flight Stewardess HAMFAZLIN SHAM BINTI MOHAMEDARIFIN Malaysia F
14 Flight Stewardess NUR SHAZANA BINTI MOHAMED SALLEH Malaysia F
15 Flight Stewardess ANGELINE PREMILA RAJANDARAN Malaysia F